
My Research
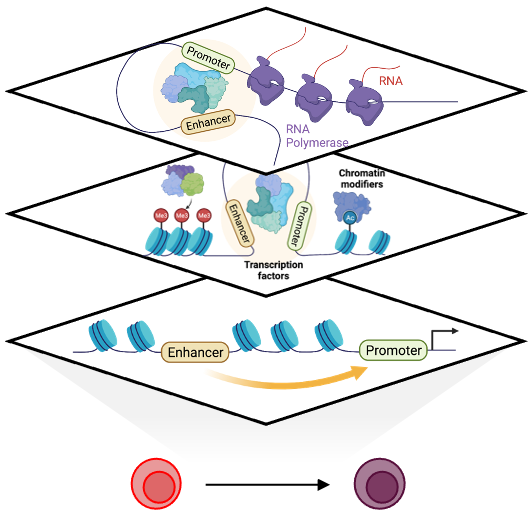 The temporal regulation of gene expression by transcription factors, chromatin modifiers and cis-regulatory elements is central to establish cellular identity and function. Understanding this regulatory logic is critical for deriving select cell types in vitro for translational applications. The human hematopoietic system has long been a model system and an important source for adoptive cell therapies, yet, our understanding of the regulatory mechanisms that elicit commitment toward distinct hematopoietic lineages is continuously evolving.
The temporal regulation of gene expression by transcription factors, chromatin modifiers and cis-regulatory elements is central to establish cellular identity and function. Understanding this regulatory logic is critical for deriving select cell types in vitro for translational applications. The human hematopoietic system has long been a model system and an important source for adoptive cell therapies, yet, our understanding of the regulatory mechanisms that elicit commitment toward distinct hematopoietic lineages is continuously evolving.
In my PhD research, I studied multiple layers of gene regulation that underly dynamic cellular processes, and sought to modulate these processes to direct differentiation to cell types of interest. My research spans building new technologies to basic science inquiry, as well as multiple fields, including hematopoiesis, synthetic biology, bioinformatics, and chromatin biology.
Live-cell transcriptomics via virus-like particles
Measuring the transcriptional states of living systems provides insight into their biological status and associated molecular mechanisms. However, non-destructive methods capable of retrieving and monitoring transcriptome-wide RNA information at multiple time points from the same living cells have yet to be developed. We overcame this limitation with a genetically encodable technology by repurposing the Gag polyprotein from murine leukemia virus (MLV) to create engineered virus-like particles (VLPs) that can export RNA molecules from cells in a process we term cellular “self-reporting”. VLP-captured RNAs were highly reflective of the host cellular transcriptome across a diverse array of human cell lines, in addition to pluripotent stem cells and primary patient fibroblasts. Furthermore, we engineered Gag with RNA-binding domains to tailor the repertoire of RNAs that are packaged in VLPs and to enrich for specific RNA transcripts. Pseudotyping VLPs with epitope-tagged VSV-G proteins enabled multiplexed self-reporting from cellular co-cultures, where each cell type was marked by a unique epitope and immunoprecipitation of each epitope-tagged VLP could deconvolve each cell type’s VLPs from the cellular media. Finally, we demonstrated that live-cell self-reported transcriptome measurements faithfully detect temporal changes in gene expression programs during induced pluripotent stem cell differentiation to mesoderm progenitors. Self-reporting of transcriptional states with engineered VLPs enables tractable, live-cell transcriptome-wide measurements from the same biological samples over time, and we envision this technology to facilitate further investigation into temporal cellular dynamics.
Transposable element regulation of hematopoietic lineage decisions
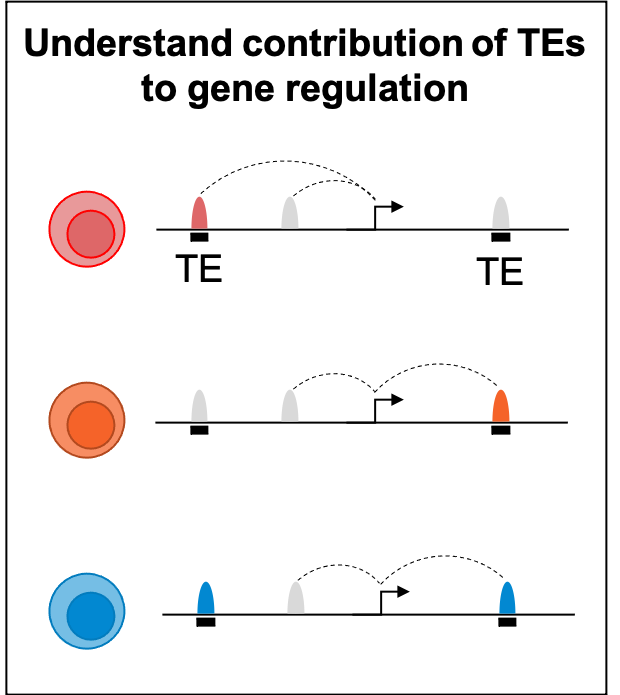 The hematopoietic system is a paradigm of stem cell biology and the focus of decades of inquiry into the molecular wiring that governs differentiation. However, the role of transposable elements (TEs) remains an underexplored layer of genetic regulation. Accumulating evidence suggests TEs were co-opted as cis-regulatory elements that shape gene regulatory networks. Moreover, TEs are regulated by epigenetic machinery and thus potently contribute to local chromatin environments. We hypothesized that TEs impart a regulatory architecture through genetic enhancers and chromatin states to guide human hematopoietic fate decisions. We generated a comprehensive atlas of enhancer-gene regulation using the Activity-by-Contact (ABC) model on all major human hematopoietic cell types amounting to 3.7 million enhancer-gene predictions. TEs were disproportionately enriched in ABC enhancers of lymphoid cells compared to other lineages, harboring lineage-specific TF motifs. TE subfamilies were transcriptionally upregulated during lymphoid differentiation, whereas the expression of TE repressive machinery, such as H3K9 methyltransferases and NuRD complex members attenuated, implying that epigenetic regulation of TEs is concomitant with lymphoid fate specification. We systematically interrogated genes involved in TE regulation within umbilical cord blood HSPCs and assessed lymphoid lineage commitment, identifying chromatin modifiers that when chemically or genetically perturbed resulted in a robust lineage shift from T to NK cells during in vitro differentiation. Genetic knockouts induced distinct NK progenitor states, identified by single cell RNA/ATAC-Seq, and derepressed TEs harbored NK-relevant TF motifs, suggesting that TE derepression facilitates NK lineage choice. Characterization of the resultant NK cells revealed distinct properties, exhibiting either proinflammatory state characterized by upregulation of interferon pathway genes, or an enhanced cytotoxic CD16+ phenotype. This work underscores the regulatory contributions of TEs and chromatin machinery on human hematopoietic lineage decisions and highlights how modulating TEs could be leveraged for hematopoietic cell engineering.
The hematopoietic system is a paradigm of stem cell biology and the focus of decades of inquiry into the molecular wiring that governs differentiation. However, the role of transposable elements (TEs) remains an underexplored layer of genetic regulation. Accumulating evidence suggests TEs were co-opted as cis-regulatory elements that shape gene regulatory networks. Moreover, TEs are regulated by epigenetic machinery and thus potently contribute to local chromatin environments. We hypothesized that TEs impart a regulatory architecture through genetic enhancers and chromatin states to guide human hematopoietic fate decisions. We generated a comprehensive atlas of enhancer-gene regulation using the Activity-by-Contact (ABC) model on all major human hematopoietic cell types amounting to 3.7 million enhancer-gene predictions. TEs were disproportionately enriched in ABC enhancers of lymphoid cells compared to other lineages, harboring lineage-specific TF motifs. TE subfamilies were transcriptionally upregulated during lymphoid differentiation, whereas the expression of TE repressive machinery, such as H3K9 methyltransferases and NuRD complex members attenuated, implying that epigenetic regulation of TEs is concomitant with lymphoid fate specification. We systematically interrogated genes involved in TE regulation within umbilical cord blood HSPCs and assessed lymphoid lineage commitment, identifying chromatin modifiers that when chemically or genetically perturbed resulted in a robust lineage shift from T to NK cells during in vitro differentiation. Genetic knockouts induced distinct NK progenitor states, identified by single cell RNA/ATAC-Seq, and derepressed TEs harbored NK-relevant TF motifs, suggesting that TE derepression facilitates NK lineage choice. Characterization of the resultant NK cells revealed distinct properties, exhibiting either proinflammatory state characterized by upregulation of interferon pathway genes, or an enhanced cytotoxic CD16+ phenotype. This work underscores the regulatory contributions of TEs and chromatin machinery on human hematopoietic lineage decisions and highlights how modulating TEs could be leveraged for hematopoietic cell engineering.
iPS-derived lymphoid cells
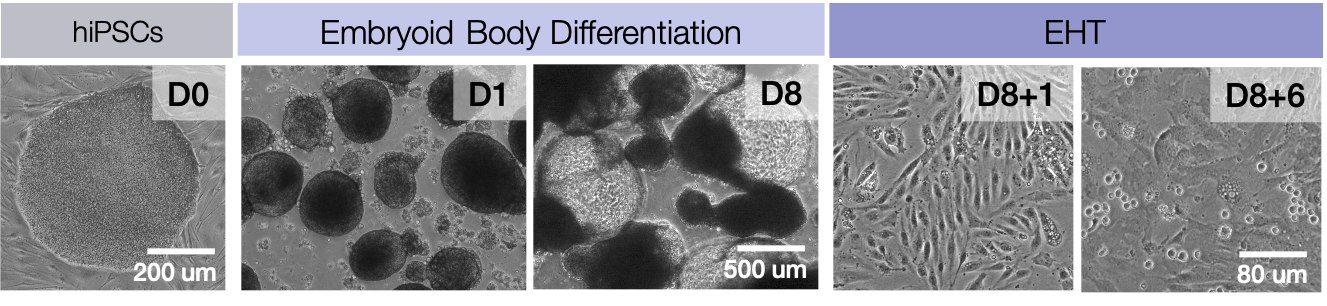
The first hematopoietic stem cells (HSCs) are generated from hemogenic endothelium during a developmental process termed, endothelial-to-hematopoietic transition (EHT). Intense scientific investigation has been devoted to understanding the molecular mechanisms of HSC ontogeny in an effort to recapitulate embryonic development to in vitro derive engraftable HSCs from induced pluripotent stem (iPSCs). In an effort to identify molecular deficiencies in current iPS differentiation protocols, we interrogated the transcriptional and chromatin transitions during EHT. We profiled hemogenic endothelium and hematopoietic progenitor populations using RNA-Seq and ATAC-Seq to discern the transcriptional and accessible chromatin landscapes over a multi-day time course of in vitro EHT. Joint analysis of chromatin and transcriptional data identified molecular discrepencies of iPSC-derived hematopoietic progenitors compared to bona fide HSCs. Infered regulatory networks nominated genes for modulation during iPSC differentiation. We experimentally identified chromatin modifiers that when manipulated during EHT enhance commitment toward lymphoid lineages, particularly T cells. Taken together, this study provides a rich molecular resource of a human model of EHT, and further advances the ability to generate therapuetically-relevant hematopoietic cell types from iPSCs for adoptive cell therapies.
About the Cover Image
Human HT1080 cells were engineered to express GFP and additional genetic machinery to enable the cells to “self-report” their transcriptional states in virus-like particles.